Cite
Chen YC. Beware of docking! Trends Pharmacol Sci. 2015 Feb;36(2):78-95. doi: 10.1016/j.tips.2014.12.001. Epub 2014 Dec 24. Erratum in: Trends Pharmacol Sci. 2015 Sep;36(9):617. PMID: 25543280.
编前语
well,我知道你们此时充满疑惑,我刚刚不久之前刚刚写过一篇文章关于对接的综述,这次便是这个题目,就仿佛是在打自己的脸。但本质上某个领域有优势自然也有缺点,我们远未达到可以依靠某个单一技术手段去解决一个科学问题或者工程的时代。关于分子对接,上篇的综述主要是讲了其应用以及方法,这次,我们来看看这项技术手段的缺点,作为互补,以便更明确的展示DOCKING的全貌,以及他究竟能做些什么,应该注意什么。

摘要
分子对接经常被用于虚拟筛选或者先导化合物优化过程中。在过去十年中,分子对接相关的论文数量急剧增加。然而,在进行对接研究时,有许多问题需要考虑。经常出现的问题,如目标蛋白的错误结合位点、使用不合适的小分子数据库进行筛选、对接姿势的选择、较高的对接分数但在分子动力学(MD)模拟中失败,以及对复合物是抑制剂还是激动剂缺乏明确性。在执行对接之前,这些问题应该引起注意和并被考虑到。一些论文展示了丰富且全面的生化实验,但最终结果只是一个简单的对接图。此综述提供了一些证据表明,有可能对接得分很高,但对接结果是存在问题的。在某些情况下,对接精度甚至可以从0%变为92.66%。因此,请认真对待。
对接本质是基于结构的药物筛选(SBDD)
请注意是基于结构的对接,并不是基于对接的结构
从20世纪60年代开始,随着物理学、化学、信息技术、生物化学和计算机的发展,分子对接已经成为一种强有力的工具和基本技术,不仅在药物筛选方面,而且在蛋白质-蛋白质相互作用和纳米材料等方面都显示出了巨大的潜力。目前计算机辅助药物设计(CADD)领域主要是将小分子对接到大分子(特别是蛋白质靶点上),其应用案例逐年增加。在现代CADD中,基于结构的药物设计是必不可少的,大多数大型制药公司都有这个部门。许多商业药物是直接使用CADD方法进行设计的。毫无疑问,在理解药物作用的分子机制上,对接技术是一项非常重要的科学进步,特别是当三位顶尖计算科学家获得2013年诺贝尔化学奖时。
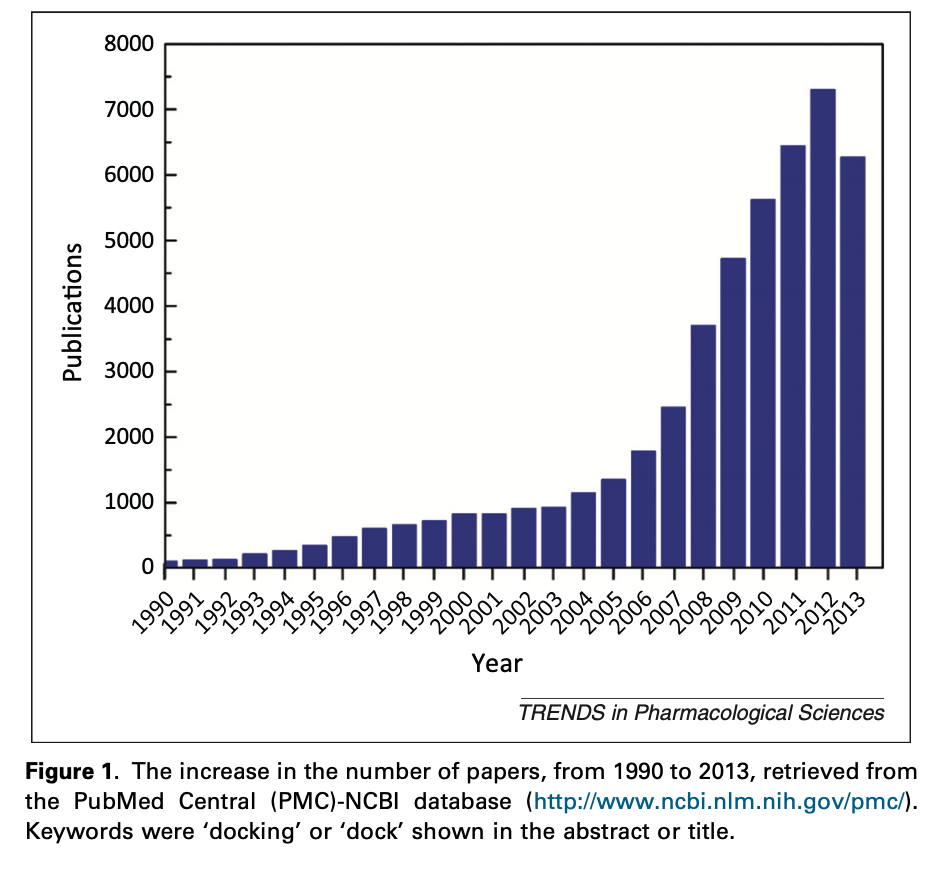
蛋白质-配体或蛋白质-蛋白质对接是一种计算技术,用于预测配体与蛋白质受体结合时的取向。多数情况下,可以选择最佳的“结合亲和力”配体进行进一步的生物化学实验。因为对接很简单,设备要求也很低(甚至在个人电脑上也能工作),对接相关的论文在过去十年里急剧增加。然而,我们能否相信这些对接研究的结果?本文对相关领域进行了调查,指出了优势和劣势。评估表明,精确性是对接研究的一个主要问题,因为如果不能精确地对接,那么这些论文就没有什么价值。
即使在知名期刊上,也能找到可疑的对接结果。常见的问题有:靶蛋白的结合位点不准确,使用不合适的小分子数据库进行筛选,对接姿势(POSE)的选择,高对接分数(结合亲和力)但在MD模拟中失败,缺乏对化合物是抑制剂还是激动剂的解释,或者对接结果与结果不一致生物测定。一些情况常常出现在一些顶级期刊上,这些期刊显示了极好的生物测定,但只有一个简单的对接图。对接结果在解释这些问题时应该引起警惕和关注。虽然有些论文通过比较对接前后配体的结合姿势差异来表明对接结果的准确性很高,但作者提出了一些观点,表明对接仍然可能存在问题。在某些情况下,对接精度甚至可以从0%变为92.66%。
对接算法和程序
Well,基本关于对接的论文都绕不开此项的讨论,我上一篇可能更详细,但是此篇主要偏向于对接程序的总结,总而言之,互有偏重。
对接的最初概念来自于“锁钥”概念,但将“钥匙”(配体)与“锁”(受体蛋白)匹配的算法多种多样。下表列出了对接程序、对接web服务器、屏幕软件和屏幕web服务器的最新发展,从中我们可以看出,近年来新算法的数量不断增加。如果我们进一步分析所有对接软件,我们可以看到最常用的对接程序是Autodock和GOLD。这并不意味着Autodock或GOLD比其他对接程序更精确,它们只是更受欢迎和知名。它们的高引用率可能是因为这些程序是免费的,并且比最近的其他对接程序创建得更早。目前,有一种新的蛋白质结构预测算法Rosetta(http://boinc.bakerlab.org/),也得到了高度评价。
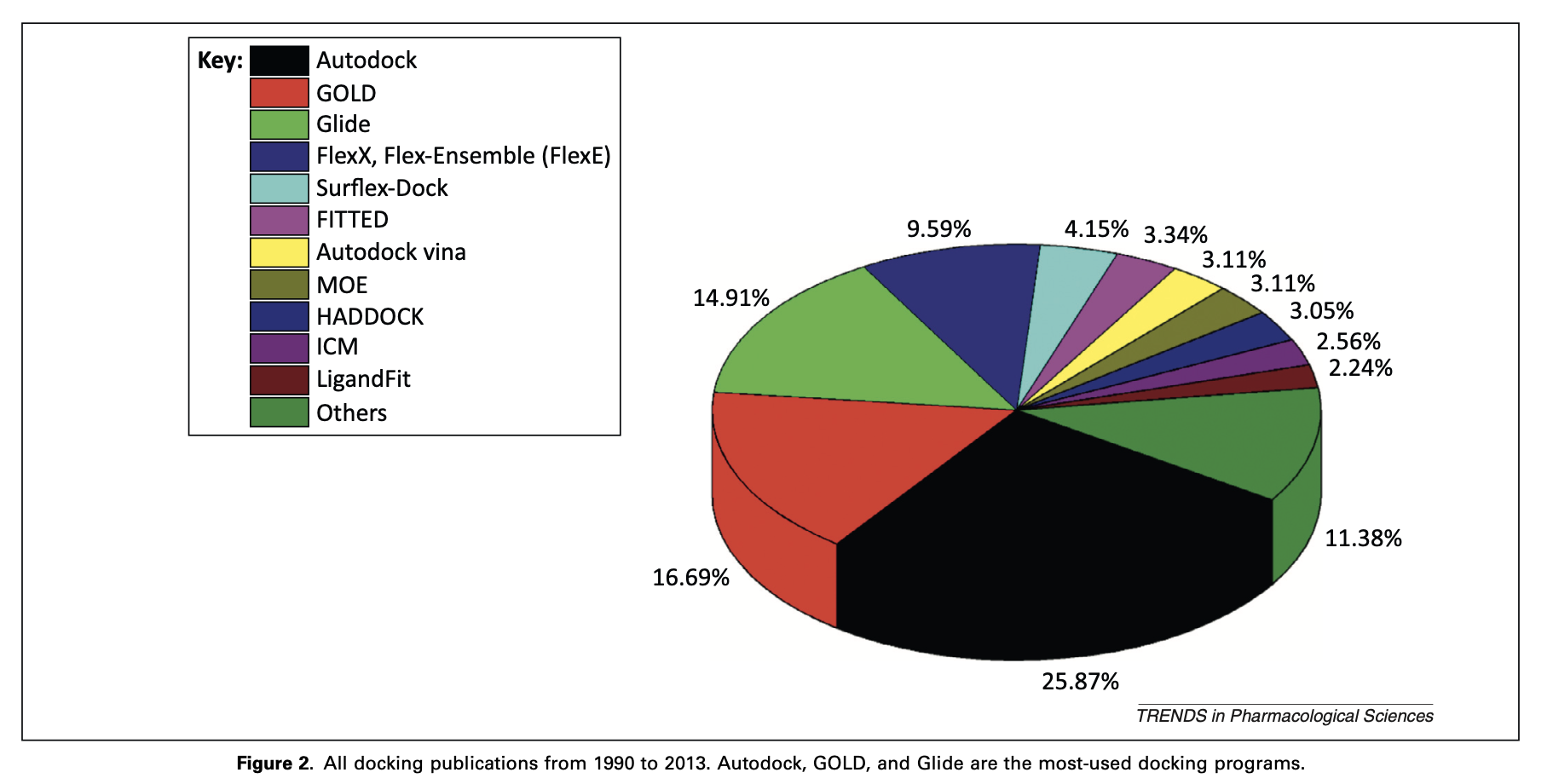
尽管程序多种多样,但每个对接程序的算法必须在速度和精度之间取得平衡。对接的算法也因评分函数的不同而有差异。结合亲和力(Binding affinity)通常被认为是一个最优先考虑因素。有不同的对接程序可以供用户根据自己的特殊要求进行选择。目前,对接算法包含了了基于结构的药物设计(SBDD)的不同方面,如基于片段的药物设计、柔性对接、水,特定pH值溶剂下对接。例如,如果我们需要从一个数据库中筛选超过10000种化合物,那么柔性对接可能不是一个好的选择,除非我们有一台强大的服务器。如果我们只需要在特定的蛋白质结合位点、特定的pH值、水或溶剂化条件下对接一些化合物,那么柔性对接程序可能是一个不错的选择。因此,对接程序的选择取决于您拥有的硬件类型以及正在筛选的数据库的规模。根据本文作者经验,将药物从CADD转化为临床是比较困难的。例如,通过对接从中医药数据库中筛选出的一些top hits在最终生物活性测试中失败。
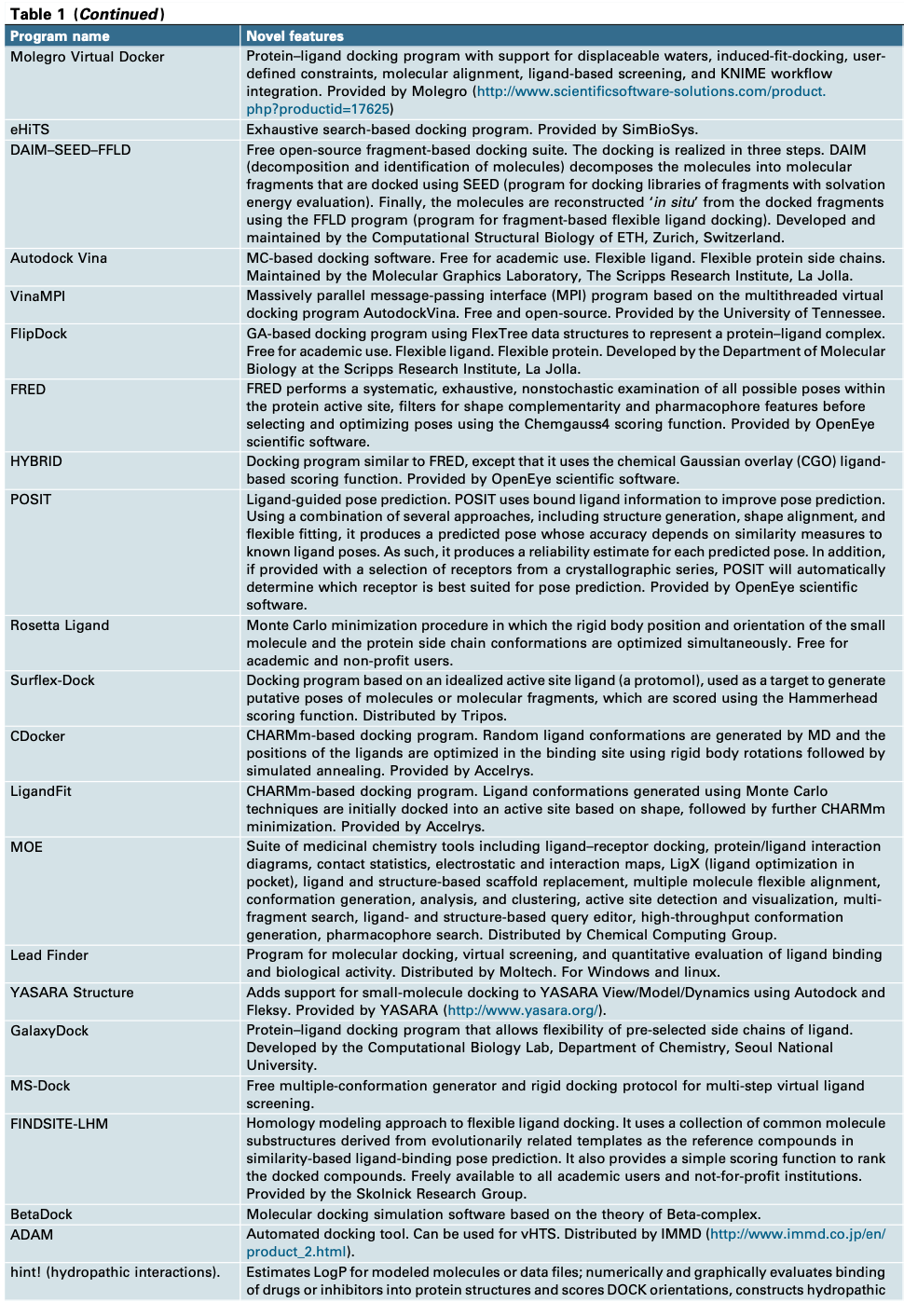
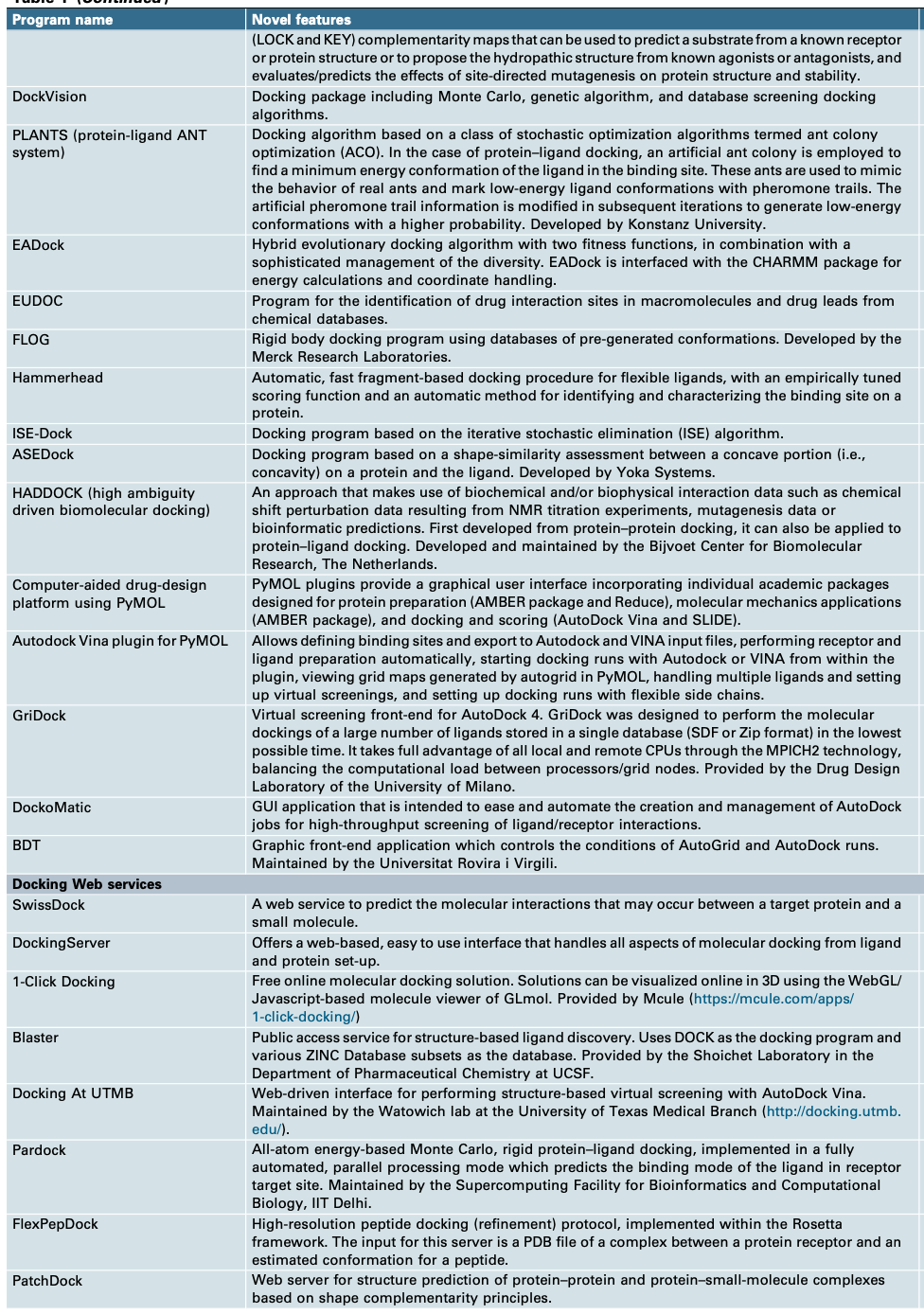
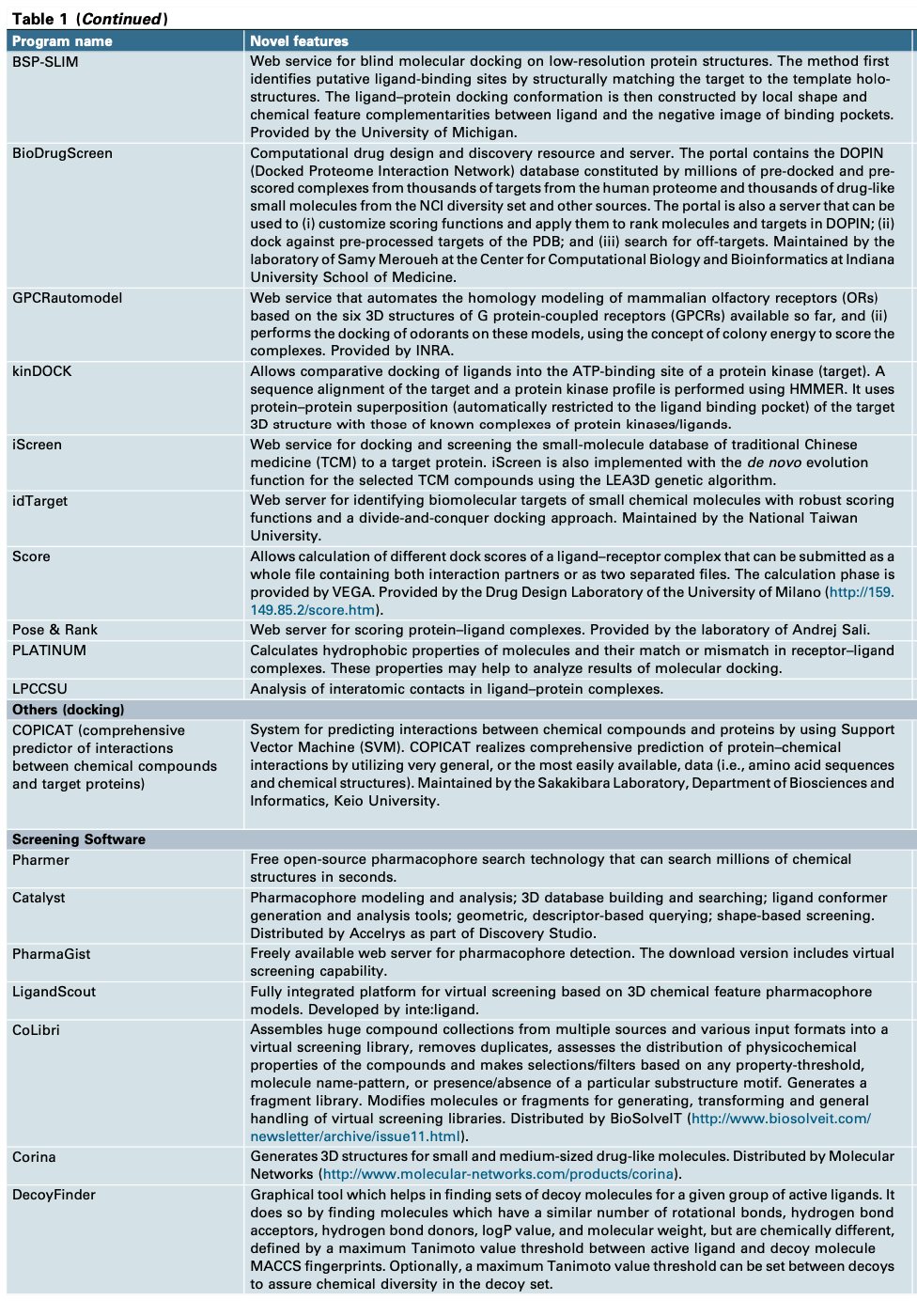
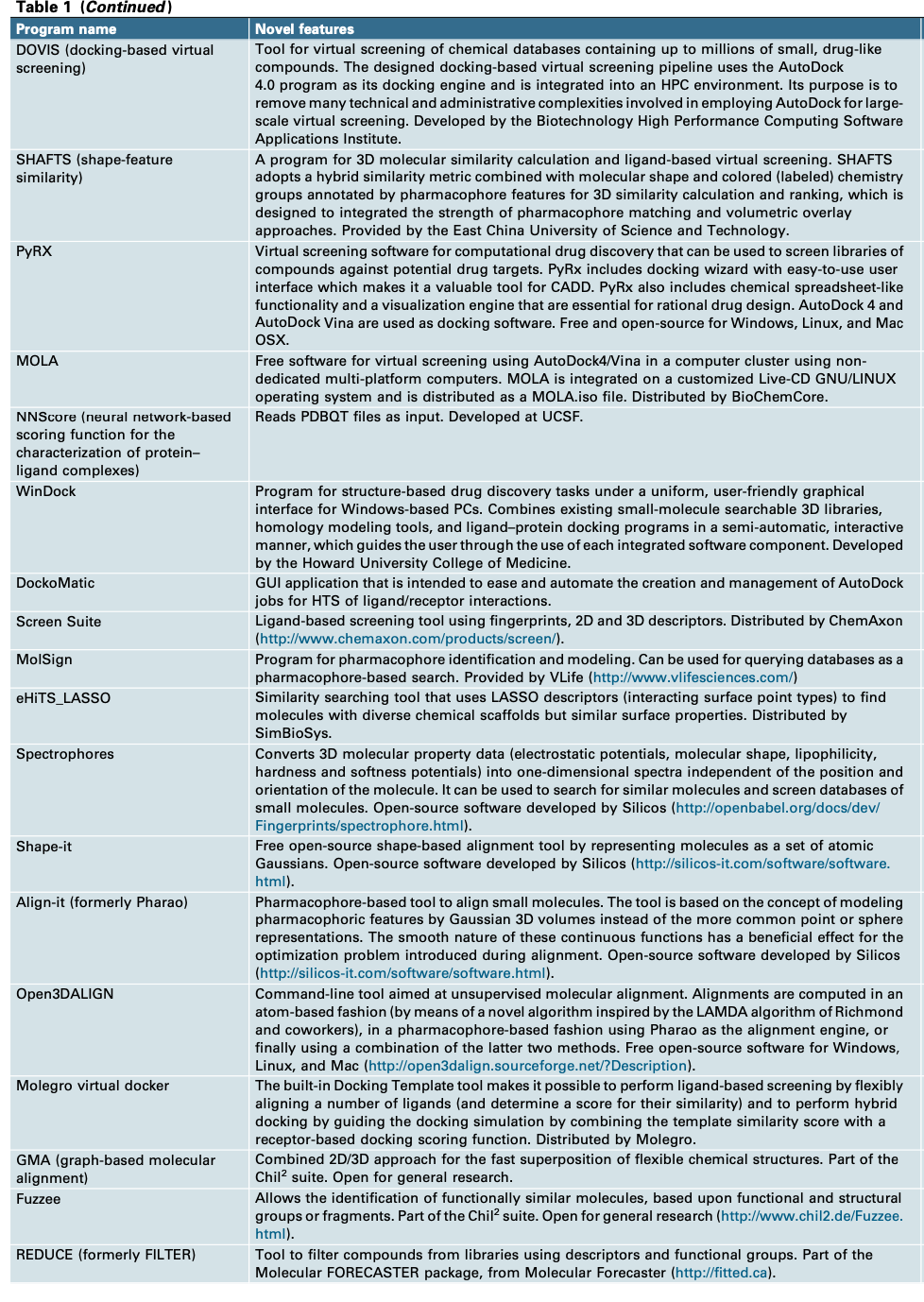
Table
最新的软件和网络服务器,基本都总结自此网站:http://www.Click2Drug.org
这个网址还是不错的,有时间单独出一篇
瑞士生物信息学研究所(SIB-swissinstituteofbioinformatics)提供了一系列计算机辅助药物设计软件和基于结构和配体计算的web服务
table 我就不翻译了,大家自己去需要的部分进行探索就好
然而,如果hits首先来自生物测定,例如,如果是通过自动化机器人技术、数据处理和体外软件利用高通量筛选(HTS)中筛选出来的——那么就更容易转移到体内。也更容易解释潜在的生物学机制。在对接研究中,谨慎是很重要的。下面列举了一些重要的因素,开发人员在进行对接时应该牢记这些影响因素。
只是列举,不是全部
蛋白结构可用吗?可信吗?
在尝试对接之前,需要做大量的准备工作。这包括使用配体和蛋白质的能量最小化来确定稳定的三维结构。
当然此项属于部分重要操作,但不是必须操作,毕竟局部最小值不等于全局最小值。
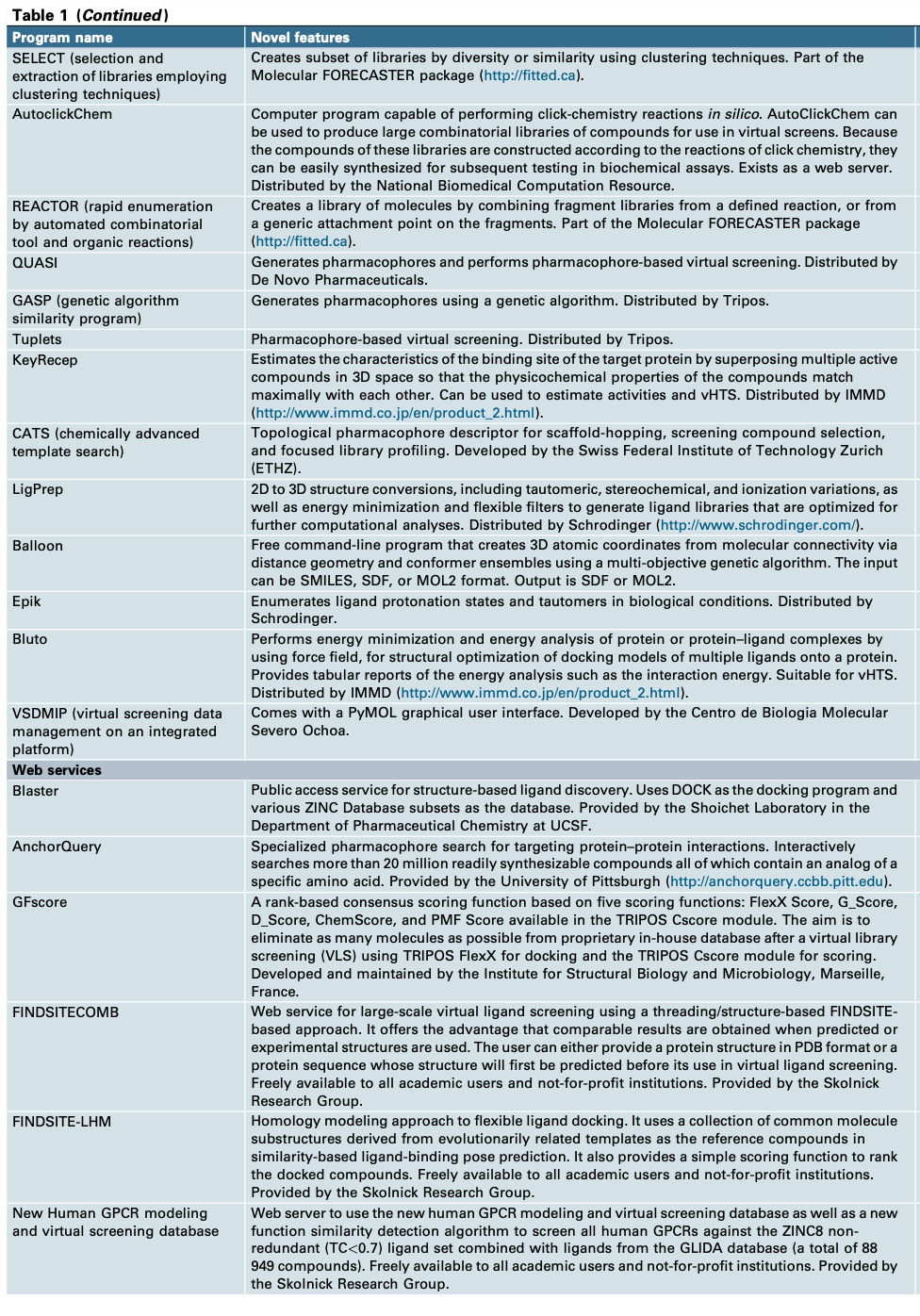
这项准备工作可以在下面的流程图中看到。如图所示,目标蛋白质序列应该可知,然后蛋白质结构应该可用,可以从同源性建模、从头建模或从蛋白质数据库(PDB)下载(http://www.rcsb.org/pdb)下载蛋白结构. 如果蛋白质和配体准备好了,就可以进行对接。现阶段的一个主要问题是蛋白质结构是否可用,是否可靠。如果蛋白质结构是从PDB数据库中下载,必须注意分辨率和获得蛋白质结构的条件。不同的温度,可以获得不同的蛋白结构。 我们可以直接利用这些蛋白质结构而不需要进一步的能量最小化或MD和聚类吗?MD模拟得到的蛋白质结构的精度会受到力场精度的影响。如果在PDB中没有蛋白质结构,那么难度将急剧上升。蛋白质结构预测的方法很多。此外,许多不同类型的验证方法被用来提高结构预测的准确性,例如著名的Ramachandran plot(拉氏图)。
柔性对接还是刚性对接
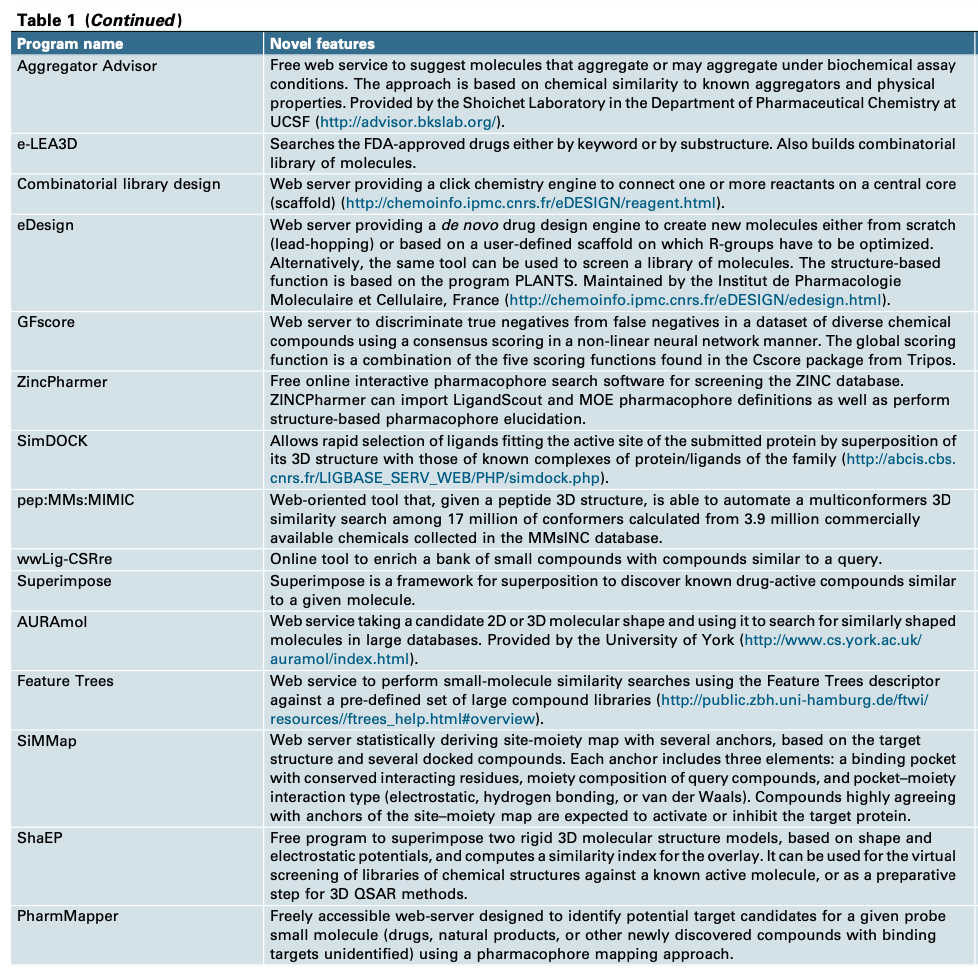
刚性对接是指蛋白质和配体构象固定,使键角或长度不变。柔性对接,允许构象的变化,今天被广泛使用的是半柔性对接,尽管它需要更多的时间和计算资源。其他对接方法包括改变溶剂化、改变pH值以及在有无水的情况下对接。用户可以根据其计算硬件和目标蛋白质的特性选择不同的对接模式。对接大致可以分为四种模式(从原理上来说):锁和钥匙、构象异构、诱导契合和构象选择。从下图可以看出,似乎只有诱导契合模式会改变口袋的形状。然而,根据MD实验的模拟,配体进入结合口袋有时会改变结合位点的形状。当配体结合时发生构象变化时,刚性对接程序将不能提供有用的结果。这表明,将蛋白质结构构象固定的对接结果可能存在错误。
如何去选择最佳的打分函数
有许多评分函数,例如,LigScore1、LigScore2、PLP1、PLP2、PMF、Dock Score、Jain、Ludi和结合能。如何选择合适的打分函数来挑选出正确的结合姿势和可能的hits?这是一个很难回答的问题。理论上,较低的吉布斯自由能表明蛋白质-配体复合物更稳定。然而,这些打分函数是针对用户的不同需求而设计的。很难确定哪种评分函数适合不同的靶蛋白。例如,我们之前已经讨论了每个打分函数的准确性,这是使用IC50与每个评分函数的回归得出的结果。我们还提出了加权打分函数,并表明其精度优于一致性打分。加权打分比一致性打分函数更准确也就不足为奇了。一致性评分允许用户选取多个或所有评分函数来评估。每个评分函数在共识评分函数中的权重相等。与此相反,加权评分是基于它们的回归系数。在该算法中,回归系数越高,权重越大。因此,加权评分比普通的一致性打分更准确。
然而,加权评分函数必须有足够的关于化合物IC50的信息,以便从评分函数中获得预测IC50的公式。它比一致性得分更精确,但不太实用,因为在大多数筛选情况下,很难获得足够的IC50数据。如果一个靶蛋白有数百个不同的配体具有不同的IC50,那么我建议使用加权评分。然而,根据我们的经验,很难从一篇论文中收集到如此多的生物活性数据。如果从不同的论文中收集生物活性数据,它们可能会因实验室环境以及protocol的不同而有所不同。普通的一致性评分避免了这个问题,因为它不需要生物活性数据。选择一致性打分或加权得分涉及到用户的所处的环境。显然,很难获得如此庞大的生物活性数据,然后用它得到最优权重评分函数,这需要大量的工作。
加权打分函数
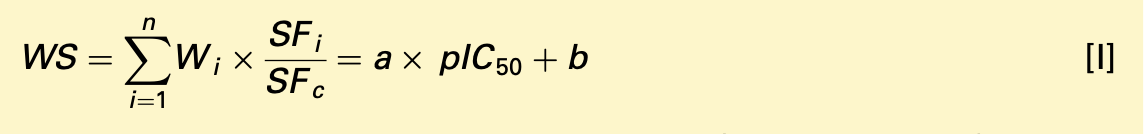
其中打分函数的数目;pIC50=-log(IC50);SFc是对照打分函数的值;SFi是打分函数的值;Wi是权重。方程中的SFc用于归一化加权方程。
打分函数的精确度
决定使用什么样的对接程序来对特定的靶蛋白进行筛选存在问题的。基本上,如果可以对生物活性与评分函数进行回归分析,那么我们就可以选择最佳的打分函数回归作为标准。准确度似乎是可以接受的,但是既然大多数hits在计算、体外或体内似乎都有很好的结合亲和力,为什么很难找到一种商业药物的先导化合物呢?首先,如果我们仅仅从PDB网站下载蛋白质-配体复合物,并从复合物中删除配体而不进一步分离纯蛋白质结构,那么蛋白质结构可能会有问题。其次,结合位点的环境是一个大问题。例如,结合位点可能位于细胞膜的内表面,因此即使配体具有较高的dock分数,它仍然必须转移到疏水性脂双层中才能到达结合域。第三,目标蛋白可能位于人体内不同的pH值。
如何知道具有较高打分的候选化合物是否可以说吗此药物时拮抗剂还是抑制剂?
如何确定一个高分化合物是一个抑制剂还是激动剂是十分困难的。许多研究仅仅使用一个对接程序,然后声称他们已经找到了激动剂或抑制剂。但是我们怎么知道这个复合体的真正机制呢?对接程序只提供计算出的结合亲和力。仅此而已,没有别的。一些论文声称,一个对接结果良好的配体表明它是一个强有力的激动剂或抑制剂。当然,在我们对生物测定进行进一步验证之前,我们不会知道它是激动剂还是抑制剂。从对接结果我们唯一确定的是配体在结合位点结合良好。因此,除非进行了其他验证,否则不建议在论文中过度解释对接结果。
对接结果与基于配体的研究不一致
有时,基于配体的研究表明,该模型是完美的,具有良好的R方(表明该模型是可靠的预测)。然后,我们可以使用这个基于配体的模型(LBDD)来预测潜在强效candidates。不幸的是,根据我们的大多数研究,大多数基于结构的结果似乎与基于配体的结果不一致。McGaughey认为,这种差异可能是因为所有的虚拟筛选方法都依赖于数据库,对于特定的靶点可能会有很大的不同。例如,在表2中,control化合物(T2384)具有非常高的对接分数,因此基于不同算法的LBDD模型也有非常高的预测活性,包括多元线性回归(MLR)、支持向量机(SVM)和贝叶斯(BNT)。然而,其他化合物也有很高的预测活性,但dock分数很低。由表2可知,对接结果显示结合能最低的对照化合物(D71904)同样在基于配体的预测(MLR和SVM)上表现出的活性也很低。相比之下,其他化合物在dock上表现较差,但在基于配体的预测(MLR和SVM)上活性很高。在表3中,较差的dock得分也显示了MLR、SVM和BNT的高预测活性。然而,具有讽刺意味的是,control化合物(SAHA)具有非常高的dock分数,但是MLR和BNT的预测结果非常差。
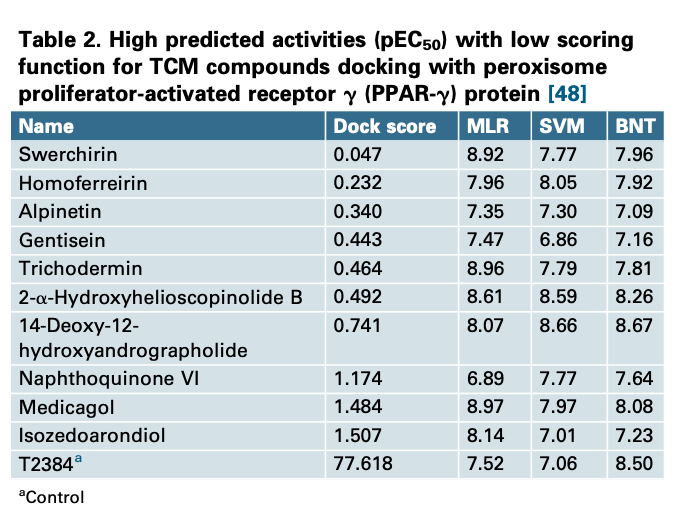
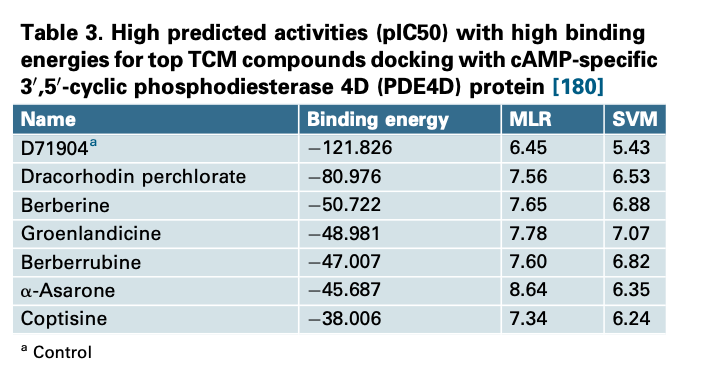
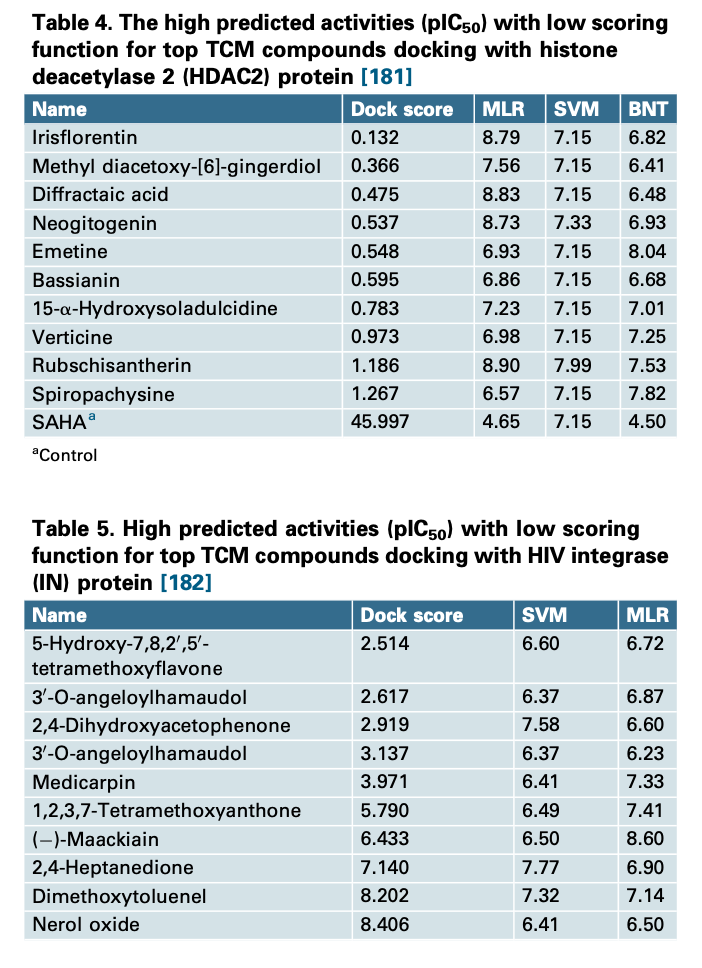
从表4可以看出,基于配体的研究(MLR、SVM和BNT)的预测活性非常高,因此表明这些化合物应该是有效的。然而,他们也表现出很差的dock分数。表5也显示了预测活性很高,但dock得分很低(方框2)。上述问题在CADD过程中并不少见,但很少出现在已发表的论文中。在他们的论文,作者把最好的数据与高dock分数结合在了一起。然而,最好的hits可能无法从最高的dock得分中找到。从我们过去的研究中,从这样的筛选中获得的前10个最有效的配体通常在真正的生物测试中失败。然而,前100名的配体往往在体外实验中显示出了good hits。这表明,虽然最高的对接分数可能不会提供最好的线索,但总的来说,对接的预测似乎没有那么糟糕。可以肯定的是,对接是基于理论计算算法,并肯定优于随机生化实验测试。

对接结果与MD结果不一致
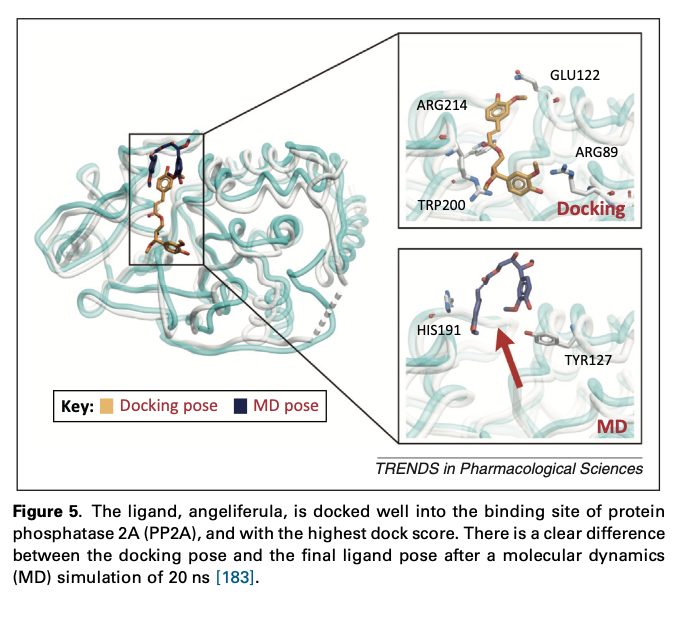
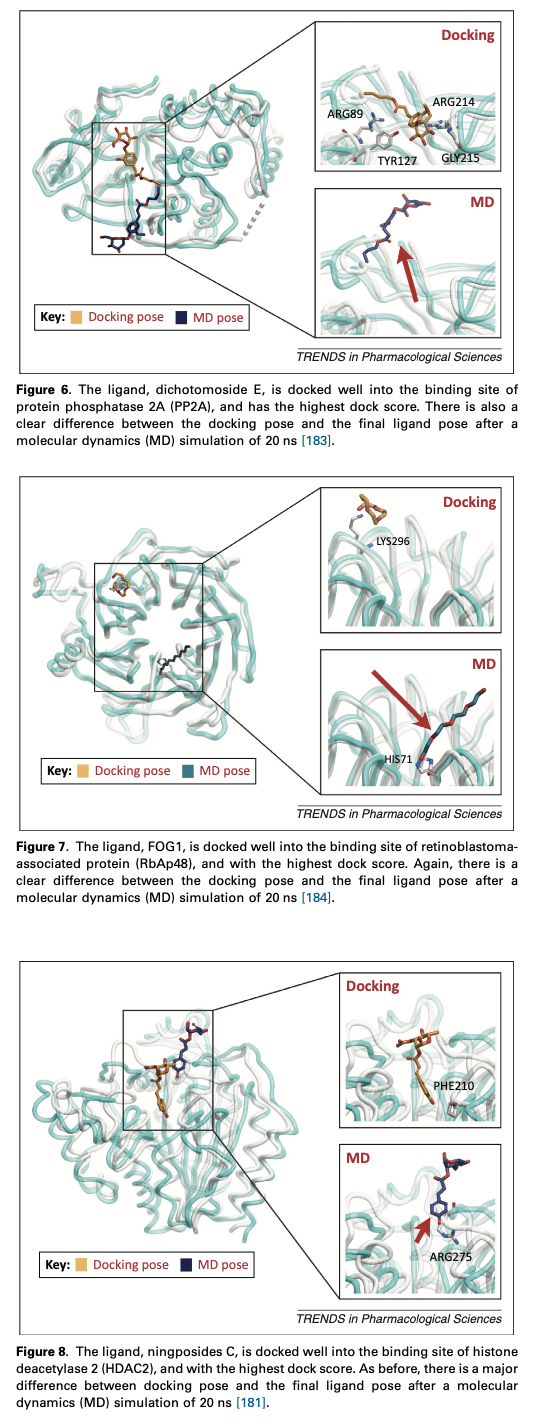
从PubMed或Web of Science数据库检索到的对接论文大多缺乏MD模拟的进一步验证。对接是一种可以计算蛋白质-蛋白质或蛋白质-配体复合物结合亲和力的工具。分析一个300 kDa的蛋白质和50-300 Da的配体结合结构通常只需要1-10秒。换句话说,对接是高通量筛选配体数据库的理想工具。MD模拟也是计算复合物构象变化“运动”的一种技术。对接和MD最大的不同时时间。对接只考虑结合能或亲和力。相比之下,MD强调了结合构象随时间的变化,与对接实验相比,MD增加了每次筛选的时间。在图5中,配体(当归)与蛋白磷酸酶2A(PP2A)的结合位点对接良好,对接得分最高。然而,对接所获得的姿势与经过20 ns的MD模拟后的最终姿势之间存在巨大差异。同样的问题可以在图6-8中看到。MD模拟也说明了配体可能“飞”出结合口袋的过程,表明MD模拟可能是我们从对接结果得出任何结论之前的必要验证。通常MD的模拟时间不是很长(由于硬件的限制,通常小于1ms)。因此,MD在CADD中肯定不是决定性的或必要的,但可以提供对接结果的进一步验证。
Summary

现在越来越多的研究依赖于先进的计算工具,因此对接结果问题变得越来越重要。许多专业的生物化学家在top期刊上发表了重要的研究结果,但只有一个简单的数字来验证配体与受体结合的关键残基。毫无疑问,这些论文都是非常重要的;然而,他们都缺乏MD研究的验证。并不是说这些类型的研究必须总是伴随着MD模拟的验证。然而,经验表明,单靠对接分析可能得出不准确的配体结合构象。尽管dock分数很高,有时配体会在MD模拟过程中飞走。因此,我强烈建议科学家们应该谨慎得出他们的结论,特别是如果只有一个对接或虚拟筛选结果,而没有进一步的验证。上图和下面提出了一个框架来对传统CADD进行改进。
计算机辅助药物设计(CADD)的集成框架与改进方法
CADD方案的完整流程图包括基于结构的药物设计(SBDD)、基于配体的药物设计(LBDD)、先导化合物优化,最后通过分子动力学(MD)模拟进行验证。对于SBDD,蛋白质结构可从蛋白质数据库获得(http://www.rcsb.org/pdb/home)或者用同源建模或从头建模。预测获得的蛋白质结构应通过**profiles-3D、Ramachandran图和无序蛋白质分析软件**进行验证。如果蛋白质的结构和结合位点符合必要的标准,我们就可以进行对接。对于筛选,应提供合适的小分子数据库。研究人员还可以筛选肽数据库。
SBDD通常与LBDD相结合来筛选潜在活性化合物。LBDD的方法是建立基于生物活性或分子描述符的模型,然后预测SBDD筛选出的化合物的生物活性。最著名的基于配体的模型QSAR是基于生物活性和药效团特征的。尽管这些模型有很大的不同,但它们都有相同的目标:在没有目标蛋白结构的情况下预测生物活性。
最后,利用MD模拟进一步验证了蛋白质结合位点的最佳对接复合物。
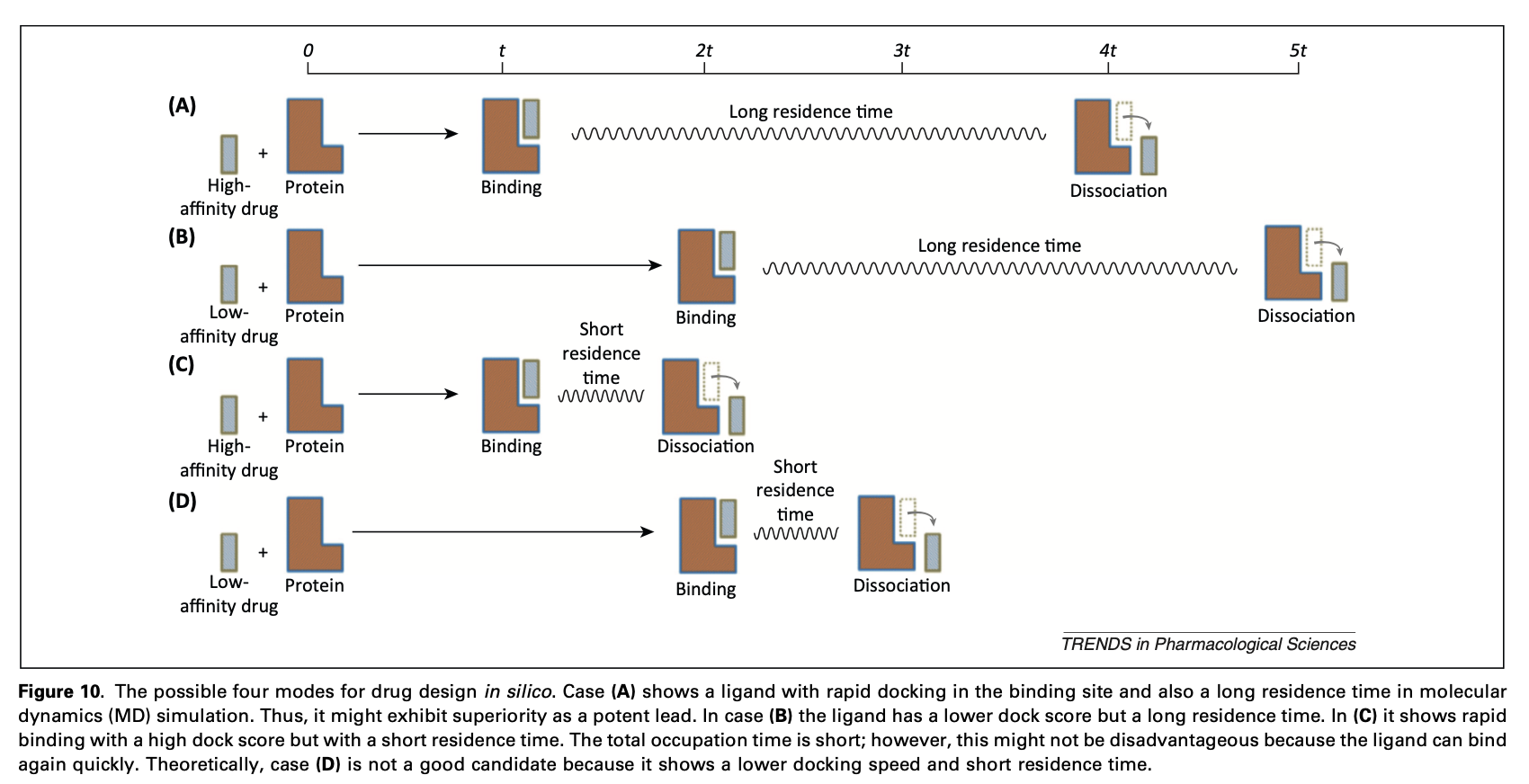
总之,药物设计原则可分为四种模式,(A)快速结合和长滞留时间(1/Koff),(B)缓慢结合但停留时间长,(C)快速结合但停留时间短,以及(D)缓慢结合和停留时间短。由于硬件和软件的限制,目前不可能使用MD来模拟复合物结构4小时或8小时知乎德变化。在未来,硬件或软件的进展可能允许MD模拟更长时间。但是,目前,MD模拟时间很短(只有几毫秒或微秒)。在本文中,MD模拟的飞走配体只是证明dock得分最高的配体并不意味着该配体是有效的先导化合物的一个证据。这是许多已发表论文中经常出现的错误。理论上,(A)模式可能是药物设计的最佳选择。然而,在实验中,我们发现超过300种商业药物和对照化合物位于(B)和(C)情境下中。辩论仍在继续。。。
Well,基本就可以分为,快结合慢解离,慢结合慢解离,快结合快解离,慢结合快解离